import pandas as pd
import chembl_downloader
from chembl_downloader import latest
from rdkit import Chem
from rdkit.Chem import Draw, AllChem
# For maximum common substructures & labelling stereocentres
from rdkit.Chem import rdFMCS, rdCIPLabeler
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.drawOptions.addAtomIndices = False
# Change to false to remove stereochem labels
IPythonConsole.drawOptions.addStereoAnnotation = True
IPythonConsole.ipython_useSVG=TrueOutline
The main goal for this post is to have a preliminary look into two of the most common groups of cytochrome P450 (CYP) inhibitors with an initial aim to look for any maximum common substructures (MCS) in these compounds if present or applicable.
TL;DR - Many pharmaceuticals have different types of rings found in their chemical structures (Taylor, MacCoss, and Lawson 2014), so it is unsurprising that the MCSes found here are mainly rings. To further explore the underlying mechanisms of actions (e.g. putative or active binding sites) of these CYP inhibitors, the structures of CYPs should be examined at the same time ideally.
Below is a general outline of the post:
- Small introduction on drug toxicology and metabolism regarding CYPs
- citing data sources for drug structural alerts in ChEMBL
- a thought on another drug metabolism data source
- Data extraction using chembl_downloader
- Data import and preprocessing
- Quick check on structural validities
- Further look into duplicated structures and stereochemistries
- quinidine
- itraconazole
- Maximum common substructures
- CYP3A4 inhibitors
- CYP2D6 inhibitors
- Some other interesting code re. MCS
- Some small findings and possible future work
- Acknowledgements
Some introductions
I initially want to work on something about drug toxicology without setting any goals or intentions on how this post will end (bit like a free-style post) and currently I feel it will be more to do with metabolism, which is also directly linked to drug toxicity, an important area not to ignore during any therapeutic drug discovery and developmental work.
This post is sort of inspired by a recent commentary that talks about “avoidome” and DMPK-related proteins to avoid (Fraser and Murcko 2024). I also happen to encounter two other blog posts (a post from D. Lowe, and another one from P. Kenny) that have provided reviews on this commentary recently with interesting view points.
In a very rough sense, three main areas have been looked at (not exhaustive) with the aim to create safer therapeutic drugs:
Structural alerts on compound substructures that are known to cause adverse drug effects or pan-assay interference compounds (PAINs)
Many have looked into structural alerts (an example repo: rd_filters). ChEMBL database has already had a cheminformatic utils web service developed that provides structural alert computations for compounds. There are most likely much more efforts than these ones.
Toxicophores in relation to human ether-a-go-go-related gene (hERG) potassium channel (related to structural alerts as well)
hERG potassium channel is also another frequently-looked-at aspect for drug toxicology due to its known effect leading to cardiac QT prolongations or more commonly known as arrhythmias (Curran et al. 1995).
CYP enzymes with the well-known ones as CYP3A4, 2D6, 1A2, 2C9 and 2C19
CYP450 enzymes play a key role in the metabolism and toxicology parts of the ADMET process of drugs. When a drug behaves like a cytochrome inhibitor, it inhibits the activity of a particular cytochrome enzyme e.g. CYP3A4 leading to a reduction of clearance of a particular therapeutic drug e.g. a CYP3A4 substrate such as apixaban, thus increasing its plasma concentration in vivo causing a higher chance of adverse effect (which in the context of apixaban, this means the poor person taking the apixaban may get excessive bleeding…).
Other useful categories involve drug-induced skin sensitisations and liver injuries and more.
My very inital naive thought is that if we can at least cover some of the drug toxicology part during drug design and discovery process, this may be able to save some resources along the way (obviously it won’t be this simple…). The main thing here is that it may still be useful and interesting to look into the relationship between CYP450 and small drug molecules - to see if there are anything worth further explorations. This post will start with the two largest groups of CYP inhibitors, so focussing on CYP3A4 and 2D6 first.
While my focus is only on a very small cohort of small molecules relating to only two CYPs, it is also worth noting that there are actually more CYPs present as well, for example, CYP1A1, 2A6, 2B6, 2C8, 2E1, 2J2, 3A5 (note: amlodipine is a moderate CYP3A5 inhibitor and will be looked at below), 3A7 and 4F2 (Guengerich 2020). The cited paper here also provides quite a comprehensive background on the history of CYP450 and their relevance to toxicities in drugs, so I won’t repeat them here.
More on structural alerts
I am only really curious about the data sources used to build these ChEMBL structural alerts, so below are some of my notes on these sources.
From ChEMBL 20, only 6 filters are present, as shown by this ChEMBL blogpost - it may appear that this blog post cites all 8 filters but in fact it only has 6. I’ve attempted to find out the sources of these ChEMBL structural alert sets, here they are:
NIH MLSMR Excluded Functionality Filters (the old link provided in the old ChEMBL blog post is no longer available, this is found via KNIME’s REOS Tagger webpage)
University of Dundee NTD Screening Library Filters (also known as “Brenk filter” in RDKit)
From ChEMBL 20 to 23, there are 8 filters in total (agreed with rd_filters’ README.md that there aren’t many documentations about this in ChEMBL, as I’ve tried also), the sources of the two additional ones are as follow:
Inpharmatica - unable to find direct source initially but this is later confirmed as private communications between ChEMBL and Inpharmatica Ltd. in the earlier days - an older ChEMBL presentation on ChEMBL 09 mentions about this, and this is also further elaborated by this paper (Gaulton et al. 2016)
SureChEMBL (old link provided by the paper (Gaulton et al. 2016) also no longer exists)
RDKit (section on “Filtering unwanted substructures”) also has another NIH filter based on two other references (Jadhav et al. 2010) and (Doveston et al. 2015). At one point I’m so confused with this NIH filter here and the NIH MLSMR one above… they are actually different as different papers are cited.
RDKit also uses the above 8 filters mentioned in ChEMBL in its FilterCatalogs class currently. Brenk filter seems to be the same as the CHEMBL_Dundee one since both of them have quoted the same journal paper as reference. It’s also got a ZINC one I think. Before I get very carried away, I’ll stop searching for every structural alerts papers here as there are many in the literatures.
More on CYPs and ADMET
A bit of a sidetrack for this part (feel free to skip) as I come across a new paper online recently about using deep learning model for ADMET prediction which uses data from Therapeutics data commons (TDC). So while working on this relevant topic of CYP and ADMET (only the metabolism and toxicology parts), I just want to dig a bit deeper to see what sort of data are used by TDC.
The TDC ADME dataset, specifically the metabolism one on all five CYP isoenzymes (CYP2C19, 2D6, 3A4, 1A2 and 2C9), are all derived from a 2009 paper by Veith et al.. A closer look at this paper only seems to mention:
…we tested 17,143 samples at between seven and fifteen concentrations for all five CYP isozymes. The samples consisted of 8,019 compounds from the MLSMR including compounds chosen for diversity and rule-of-five compliance, 16 synthetic tractability, and availability; 6,144 compounds from a set of biofocused libraries which included 1,114 FDA-approved drugs; and 2,980 compounds from combinatorial libraries containing privileged structures targeted at GPCRs and kinases, and libraries of purified natural products or related structures…
If I go to its original journal paper site (the link provided was a NCBI one), there is only one additional Excel file with a long list of chemical scaffolds showing different CYP activities (no other supplementary information I can spot there). The only likely lists of compounds tested are shown in its figures 6 and 7 in the paper, where figure 7 is more relevant for drug-drug interactions. I then realise the proportions of FDA-approved drugs used and the rest of the molecules tested in this paper are also not very balanced (thinking along the line of approved drugs and non-approved drugs), and notice that what they are saying in its discussion about how they are not noticing the usual prominent activities of CYP3A4 and 2D6 in the compounds they’ve tested:
…It has been suggested that CYP 3A4 is the most prominent P450 isozyme in drug metabolism and hepatic distribution (Fig. 2b),25, 26 but the drugs in our collection do not appear to have been optimized away from this activity. There has also been speculation that CYP 2D6 isozyme plays a prominent role in drug metabolism,27 but no difference in activity was observed between diversity compounds and approved drugs for this isozyme…
I wonder if this may be due to the imbalanced set of compounds used e.g. number of FDA-approved drug (smaller) vs. number of other compounds from other libraries (larger)…
I’ve also visited FDA’s website to look at how the CYP stories are compiled (FDA link). The in vitro inhibitors and clinical index inhibitors are not completely the same across all the CYPs. There are some overlappings in CYP3A4/5 and 2D6 for sure but definitely not exactly the same across all the documented CYPs in this FDA webpage.
So back to this new paper on predicting ADMET… how likely will it be useful in real-life hit/lead ADMET optimisation projects in drug discovery settings if the data source only involves a larger portion of non-approved drugs versus a smaller portion of actual FDA-approved drugs?… It just shows that there are a lot of things to think about in the DMPK/ADMET areas within drug discovery pipelines, as ultimately this is crucial to see if a candidate molecule will proceed or not (i.e. causing toxicity or not and whether it’s tolerable side effects or adverse or even life-threatening ones instead).
Extracting data
First step here is to import the following software packages in order to retrieve and work with ChEMBL data (again).
# Latest version of ChEMBL
latest_version = latest()
print(f"The latest ChEMBL version is: {latest_version}")The latest ChEMBL version is: 34I’m using SQL via chembl_downloader to download approved drugs with their ChEMBL ID and equivalent canonical SMILES. All of the CYP3A4 and 2D6 inhibitors extracted from ChEMBL are based on the Flockhart table of drug interactions (Flockhart et al. 2021).
Note: Three other categories of medicines are not going to be looked at for now, which are the weak inhibitors, ones with in vitro evidence only and ones that are still pending reviews.
A bit about retrieving data here, the following may not be the best way to get the data, but I’ve somehow incorporated chembl_downloader into my own small piece of function code (see Python script named as “cyp_drugs.py” in the repo) to retrieve SMILES of approved drugs (other public databases may also work very well equally, but I’m used to using ChEMBL now as it’s easy to read and navigate).
Another possible way is to use get_target_sql() within chembl_downloader, e.g. using a specific CYP enzyme as the protein target to retrieve data, but it appears that there are no clear data marked to indicate the potency of CYP inhibition or induction (i.e. weak, moderate or strong) in the ChEMBL database (an example link for CYP2D6 in ChEMBL). The Flockhart table has clearly annotated each approved drug with journal paper citations so I decide to stick with the previous method.
## Main issue previously is with sql string - too many quotation marks!
# e.g. WHERE molecule_dictionary.pref_name = '('KETOCONAZOLE', 'FLUCONAZOLE')'': near "KETOCONAZOLE": syntax error
# Resolved issue by adding string methods e.g. strip() and replace() to sql query string
from cyp_drugs import chembl_drugs
# Get a list of strong cyp3a4 inhibitors
# For the story on why I also added a weird spelling of "itraconzole", please see below.
# and save as a tsv file
df_3a4_strong_inh = chembl_drugs(
"CERITINIB", "CLARITHROMYCIN", "DELAVIRIDINE", "IDELALISIB", "INDINAVIR", "ITRACONAZOLE", "ITRACONZOLE", "KETOCONAZOLE", "MIBEFRADIL", "NEFAZODONE", "NELFINAVIR", "RIBOCICLIB", "RITONAVIR", "SAQUINAVIR", "TELAPREVIR", "TELITHROMYCIN", "TUCATINIB", "VORICONAZOLE",
#file_name="strong_3a4_inh"
)
df_3a4_strong_inh.head()| chembl_id | pref_name | max_phase | canonical_smiles | |
|---|---|---|---|---|
| 0 | CHEMBL2403108 | CERITINIB | 4.0 | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... |
| 1 | CHEMBL1741 | CLARITHROMYCIN | 4.0 | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... |
| 2 | CHEMBL2216870 | IDELALISIB | 4.0 | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... |
| 3 | CHEMBL115 | INDINAVIR | 4.0 | CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](... |
| 4 | CHEMBL22587 | ITRACONAZOLE | NaN | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](... |
## Get a list of moderate cyp3a4 inhibitors
# skipping grapefruit juice as it's not quite an approved drug...
# note: amlodipine inhibits cyp3a5
df_3a4_mod_inh = chembl_drugs(
"AMLODIPINE", "APREPITANT", "CIPROFLOXACIN", "CRIZOTINIB", "DILTIAZEM", "ERYTHROMYCIN", "FLUCONAZOLE", "IMATINIB", "LETERMOVIR", "NETUPITANT", "VERAPAMIL", #file_name="mod_3a4_inh"
)
df_3a4_mod_inh.head()| chembl_id | pref_name | max_phase | canonical_smiles | |
|---|---|---|---|---|
| 0 | CHEMBL1491 | AMLODIPINE | 4 | CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1c1ccccc1Cl |
| 1 | CHEMBL1471 | APREPITANT | 4 | C[C@@H](O[C@H]1OCCN(Cc2n[nH]c(=O)[nH]2)[C@H]1c... |
| 2 | CHEMBL8 | CIPROFLOXACIN | 4 | O=C(O)c1cn(C2CC2)c2cc(N3CCNCC3)c(F)cc2c1=O |
| 3 | CHEMBL601719 | CRIZOTINIB | 4 | C[C@@H](Oc1cc(-c2cnn(C3CCNCC3)c2)cnc1N)c1c(Cl)... |
| 4 | CHEMBL23 | DILTIAZEM | 4 | COc1ccc([C@@H]2Sc3ccccc3N(CCN(C)C)C(=O)[C@@H]2... |
# Get a list of strong cyp2d6 inhibitors
df_2d6_strong_inh = chembl_drugs(
"BUPROPION", "FLUOXETINE", "PAROXETINE", "QUINIDINE",
#file_name="strong_2d6_inh"
)
df_2d6_strong_inh| chembl_id | pref_name | max_phase | canonical_smiles | |
|---|---|---|---|---|
| 0 | CHEMBL894 | BUPROPION | 4.0 | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 |
| 1 | CHEMBL41 | FLUOXETINE | 4.0 | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 |
| 2 | CHEMBL490 | PAROXETINE | 4.0 | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 |
| 3 | CHEMBL21578 | QUINIDINE | NaN | C=C[C@H]1CN2CCC1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC... |
| 4 | CHEMBL1294 | QUINIDINE | 4.0 | C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc... |
# Get a list of moderate cyp2d6 inhibitors
df_2d6_mod_inh = chembl_drugs(
"ABIRATERONE", "CINACALCET", "CLOBAZAM", "DOXEPIN", "DULOXETINE", "HALOFANTRINE", "LORCASERIN", "MOCLOBEMIDE", "ROLAPITANT", "TERBINAFINE",
#file_name="mod_2d6_inh"
)
df_2d6_mod_inh.head()| chembl_id | pref_name | max_phase | canonical_smiles | |
|---|---|---|---|---|
| 0 | CHEMBL254328 | ABIRATERONE | 4 | C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C... |
| 1 | CHEMBL1201284 | CINACALCET | 4 | C[C@@H](NCCCc1cccc(C(F)(F)F)c1)c1cccc2ccccc12 |
| 2 | CHEMBL70418 | CLOBAZAM | 4 | CN1C(=O)CC(=O)N(c2ccccc2)c2cc(Cl)ccc21 |
| 3 | CHEMBL1628227 | DOXEPIN | 4 | CN(C)CCC=C1c2ccccc2COc2ccccc21 |
| 4 | CHEMBL1175 | DULOXETINE | 4 | CNCC[C@H](Oc1cccc2ccccc12)c1cccs1 |
Initially, four categories of approved drugs are retrieved - the strong and moderate CYP3A4 inhibitors, and also the strong and moderate CYP2D6 inhibitors. CYP3A4 inhibitors are the largest cohort of all the cytochrome inhibitors known so far (based on clinical documentations).
Import and preprocess data
## When using pandas 2.2.2, numpy 2.0.0 and rdkit 2024.3.1
# (all latest major versions at the time of writing,
# note: rdkit has a latest minor release as 2024.03.4, which includes a patch for numpy 2.0)
# Seems to work as a new df is generated but with error messages shown
## Eventually using downgraded versions of pandas and numpy instead
# pandas 2.1.4, numpy 1.26.4 & rdkit 2024.3.1 work with no error messages generated
# preprocess canonical smiles
from mol_prep import preprocess
# cyp3a4 strong inhibitors
df_3a4_s_inh = df_3a4_strong_inh.copy()
df_3a4_s_inh_p = df_3a4_s_inh.apply(preprocess, axis=1)
df_3a4_s_inh_p.head(3)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL2403108 | CERITINIB | 4.0 | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | <rdkit.Chem.rdchem.Mol object at 0x135da19a0> | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | [C][C][=C][C][Branch2][Ring2][=Branch1][N][C][... | InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-... | VERWOWGGCGHDQE-UHFFFAOYSA-N |
| 1 | CHEMBL1741 | CLARITHROMYCIN | 4.0 | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | <rdkit.Chem.rdchem.Mol object at 0x135da1b60> | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | [C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran... | InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21... | AGOYDEPGAOXOCK-KCBOHYOISA-N |
| 2 | CHEMBL2216870 | IDELALISIB | 4.0 | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | <rdkit.Chem.rdchem.Mol object at 0x135da1a80> | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | [C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][... | InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-... | IFSDAJWBUCMOAH-HNNXBMFYSA-N |
# cyp3a4 moderate inhibitors
df_3a4_m_inh = df_3a4_mod_inh.copy()
df_3a4_m_inh_p = df_3a4_m_inh.apply(preprocess, axis=1)
df_3a4_m_inh_p.head(3)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL1491 | AMLODIPINE | 4 | CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1c1ccccc1Cl | <rdkit.Chem.rdchem.Mol object at 0x135da3990> | CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1c1ccccc1Cl | [C][C][O][C][=Branch1][C][=O][C][=C][Branch1][... | InChI=1S/C20H25ClN2O5/c1-4-28-20(25)18-15(11-2... | HTIQEAQVCYTUBX-UHFFFAOYSA-N |
| 1 | CHEMBL1471 | APREPITANT | 4 | C[C@@H](O[C@H]1OCCN(Cc2n[nH]c(=O)[nH]2)[C@H]1c... | <rdkit.Chem.rdchem.Mol object at 0x135da2ce0> | C[C@@H](O[C@H]1OCCN(Cc2n[nH]c(=O)[nH]2)[C@H]1c... | [C][C@@H1][Branch2][Ring2][C][O][C@H1][O][C][C... | InChI=1S/C23H21F7N4O3/c1-12(14-8-15(22(25,26)2... | ATALOFNDEOCMKK-OITMNORJSA-N |
| 2 | CHEMBL8 | CIPROFLOXACIN | 4 | O=C(O)c1cn(C2CC2)c2cc(N3CCNCC3)c(F)cc2c1=O | <rdkit.Chem.rdchem.Mol object at 0x135da2ff0> | O=C(O)c1cn(C2CC2)c2cc(N3CCNCC3)c(F)cc2c1=O | [O][=C][Branch1][C][O][C][=C][N][Branch1][=Bra... | InChI=1S/C17H18FN3O3/c18-13-7-11-14(8-15(13)20... | MYSWGUAQZAJSOK-UHFFFAOYSA-N |
# cyp2d6 strong inhibitors
df_2d6_s_inh = df_2d6_strong_inh.copy()
df_2d6_s_inh_p = df_2d6_s_inh.apply(preprocess, axis=1)
df_2d6_s_inh_p.head(3)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL894 | BUPROPION | 4.0 | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | <rdkit.Chem.rdchem.Mol object at 0x135da3530> | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | [C][C][Branch1][#Branch2][N][C][Branch1][C][C]... | InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-... | SNPPWIUOZRMYNY-UHFFFAOYSA-N |
| 1 | CHEMBL41 | FLUOXETINE | 4.0 | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3290> | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | [C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=... | InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-... | RTHCYVBBDHJXIQ-UHFFFAOYSA-N |
| 2 | CHEMBL490 | PAROXETINE | 4.0 | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3bc0> | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | [F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@... | InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-... | AHOUBRCZNHFOSL-YOEHRIQHSA-N |
#cyp2d6 moderate inhibitors
df_2d6_m_inh = df_2d6_mod_inh.copy()
df_2d6_m_inh_p = df_2d6_m_inh.apply(preprocess, axis=1)
df_2d6_m_inh_p.head(3)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL254328 | ABIRATERONE | 4 | C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C... | <rdkit.Chem.rdchem.Mol object at 0x135dd0270> | C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C... | [C][C@][C][C][C@H1][Branch1][C][O][C][C][Ring1... | InChI=1S/C24H31NO/c1-23-11-9-18(26)14-17(23)5-... | GZOSMCIZMLWJML-VJLLXTKPSA-N |
| 1 | CHEMBL1201284 | CINACALCET | 4 | C[C@@H](NCCCc1cccc(C(F)(F)F)c1)c1cccc2ccccc12 | <rdkit.Chem.rdchem.Mol object at 0x135da3e60> | C[C@@H](NCCCc1cccc(C(F)(F)F)c1)c1cccc2ccccc12 | [C][C@@H1][Branch2][Ring1][#Branch1][N][C][C][... | InChI=1S/C22H22F3N/c1-16(20-13-5-10-18-9-2-3-1... | VDHAWDNDOKGFTD-MRXNPFEDSA-N |
| 2 | CHEMBL70418 | CLOBAZAM | 4 | CN1C(=O)CC(=O)N(c2ccccc2)c2cc(Cl)ccc21 | <rdkit.Chem.rdchem.Mol object at 0x135da3b50> | CN1C(=O)CC(=O)N(c2ccccc2)c2cc(Cl)ccc21 | [C][N][C][=Branch1][C][=O][C][C][=Branch1][C][... | InChI=1S/C16H13ClN2O2/c1-18-13-8-7-11(17)9-14(... | CXOXHMZGEKVPMT-UHFFFAOYSA-N |
Images of structures
Here what I’m trying to do is to check structural validities of all the drug molecules, and one of the easiest things to do is to look at their chemical structures directly.
# moderate cyp2d6 inhibitors
Draw.MolsToGridImage(
df_2d6_m_inh_p["rdkit_mol"],
molsPerRow=3,
subImgSize=(400, 300),
legends=list(df_2d6_m_inh_p["pref_name"])
)
# strong cyp2d6 inhibitors
Draw.MolsToGridImage(
df_2d6_s_inh_p["rdkit_mol"],
molsPerRow=3,
subImgSize=(400, 300),
legends=list(df_2d6_s_inh_p["pref_name"])
)
Duplicated structures and stereochemistries
This is a small detour while checking structural validities due to the presence of two duplicated molecules, and since these two molecules consist of stereocentres, I’m just going to have a look at their stereochemistries.
quinidine
There are different stereochemistries spotted in the two quinidines shown below.
# Stereochem in RDKit
# Older approach - AssignStereochemistry() -> this is used in datamol's standardize_mol(),
# which is used in my small mol_prep.py script
# Newer approach - FindPotentialStereo()# Get 2D image of quinidine at row 3
df_2d6_s_inh_p.loc[3, "rdkit_mol"]# Get 2D image of quinidine at row 4
df_2d6_s_inh_p.loc[4, "rdkit_mol"]# Get SMILES for quinidine at row 3
df_2d6_s_inh_p.loc[3, "canonical_smiles"]'C=C[C@H]1CN2CCC1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12'# Get SMILES for quinidine at row 4
df_2d6_s_inh_p.loc[4, "canonical_smiles"]'C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12'# quinidine at index row 3
quinidine_3 = Chem.MolFromSmiles('C=C[C@H]1CN2CCC1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12')
rdCIPLabeler.AssignCIPLabels(quinidine_3)
quinidine_3Ref: Stereochemical or CIP (Cahn–Ingold–Prelog) labeller in RDKit
# quinidine index row 4
quinidine_4 = Chem.MolFromSmiles('C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12')
rdCIPLabeler.AssignCIPLabels(quinidine_4)
quinidine_4Quinidine has 4 defined atom stereocentre count as per PubChem compound summary (as one of possible references for cross-checking) - this is based on the calculation for CHEMBL1294, which is the same as the quinidine spotted at index row 4. So I’m dropping the quinidine at index row 3 for now.
# Note: old index is unchanged for now (re-index later if needed)
df_2d6_s_inh_p = df_2d6_s_inh_p.drop(labels = 3)
df_2d6_s_inh_p| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL894 | BUPROPION | 4.0 | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | <rdkit.Chem.rdchem.Mol object at 0x135da3530> | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | [C][C][Branch1][#Branch2][N][C][Branch1][C][C]... | InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-... | SNPPWIUOZRMYNY-UHFFFAOYSA-N |
| 1 | CHEMBL41 | FLUOXETINE | 4.0 | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3290> | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | [C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=... | InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-... | RTHCYVBBDHJXIQ-UHFFFAOYSA-N |
| 2 | CHEMBL490 | PAROXETINE | 4.0 | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3bc0> | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | [F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@... | InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-... | AHOUBRCZNHFOSL-YOEHRIQHSA-N |
| 4 | CHEMBL1294 | QUINIDINE | 4.0 | C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc... | <rdkit.Chem.rdchem.Mol object at 0x135da2d50> | C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc... | [C][=C][C@H1][C][N][C][C][C@H1][Ring1][=Branch... | InChI=1S/C20H24N2O2/c1-3-13-12-22-9-7-14(13)10... | LOUPRKONTZGTKE-LHHVKLHASA-N |
itraconazole
Two itraconazoles are also found with different stereochemistries.
# Get SMILES of itraconazole at index row 4
df_3a4_s_inh_p.loc[4, "canonical_smiles"]'CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](Cn6cncn6)(c6ccc(Cl)cc6Cl)O5)cc4)CC3)cc2)c1=O'# Get SMILES of itraconazole at index row 5
df_3a4_s_inh_p.loc[5, "canonical_smiles"]'CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn6)(c6ccc(Cl)cc6Cl)O5)cc4)CC3)cc2)c1=O'itracon_4 = Chem.MolFromSmiles("CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](Cn6cncn6)(c6ccc(Cl)cc6Cl)O5)cc4)CC3)cc2)c1=O")
rdCIPLabeler.AssignCIPLabels(itracon_4)
itracon_4itracon_5 = Chem.MolFromSmiles("CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn6)(c6ccc(Cl)cc6Cl)O5)cc4)CC3)cc2)c1=O")
rdCIPLabeler.AssignCIPLabels(itracon_5)
itracon_5Clearly, even if the SMILES of these two itraconzoles are not converted into RDKit molecules, we can probably tell one of them has stereochemistries and the other one is without due to the presence of “@” in the SMILES string for the one at index row 4. The output images show exactly that - one with chiral centres, where the other one doesn’t have any.
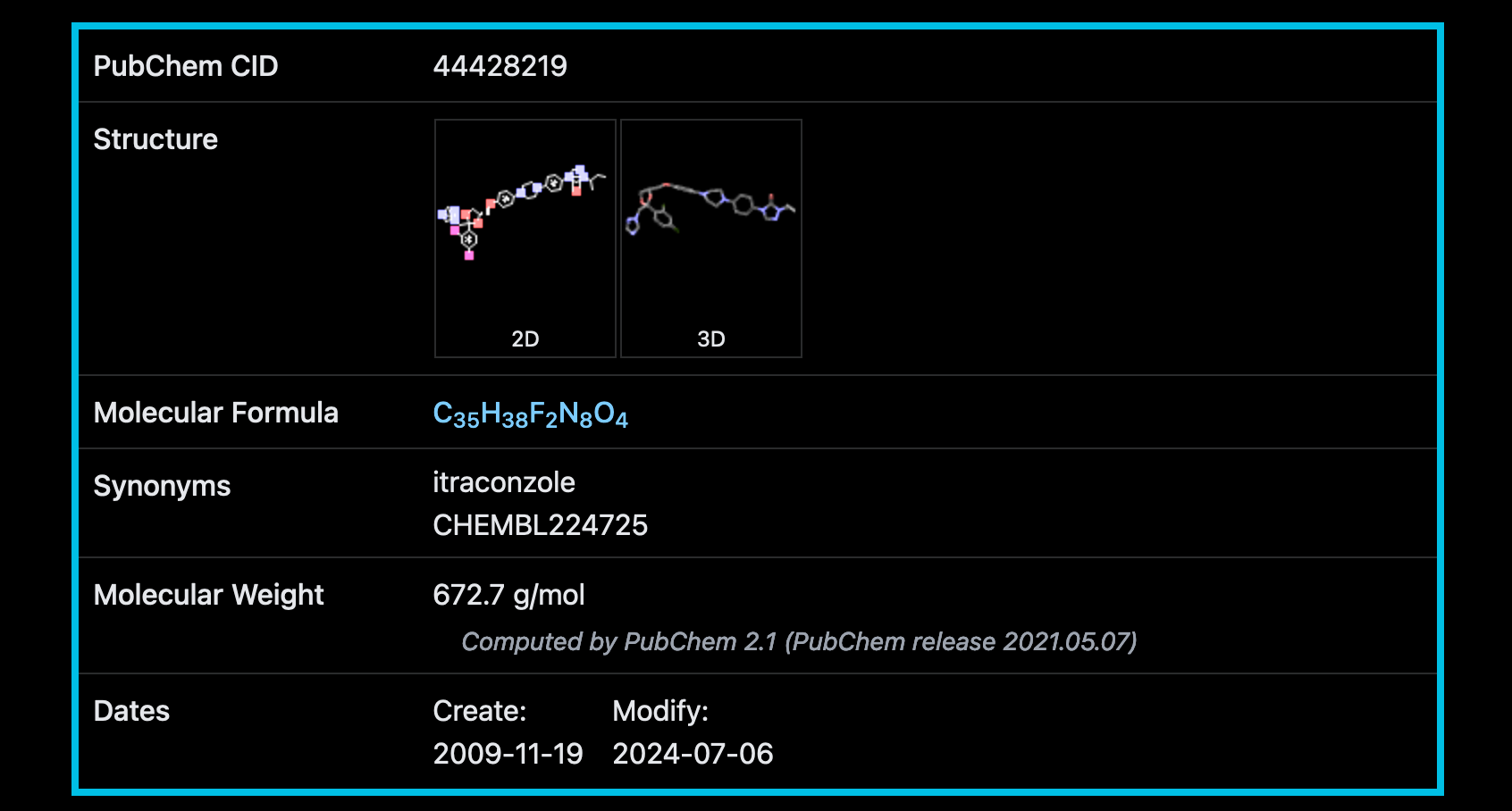
PubChem calculations have however generated different result for itraconazole. It seems it only has one defined atom stereocentre count and two undefined stereocentre counts (PubChem reference).
I’ve also noted that the two itraconzoles obtained from ChEMBL have different ChEMBL ID numbers (ChEMBL IDs: CHEMBL22587 and CHEMBL64391) to the one calculated in PubChem (ChEMBL ID: CHEMBL224725). So below I’ve looked into CHEMBL224725 first.
Then I realise that if I search for “itraconazole” directly in ChEMBL, only four entries will appear with ChEMBL IDs of CHEMBL64391, CHEMBL22587, CHEMBL882 and CHEMBL5090785, and there is no ChEMBL224725. This is all due to a small spelling error (which is most likely a typo by accident) of itraconazole - being spelled as “itraconzole”, which is also carried over into PubChem as well. I have checked again to make sure both “itraconzole” and the usual itraconazole are referring to the same chemical structure. Below are some screenshots showing the typo. 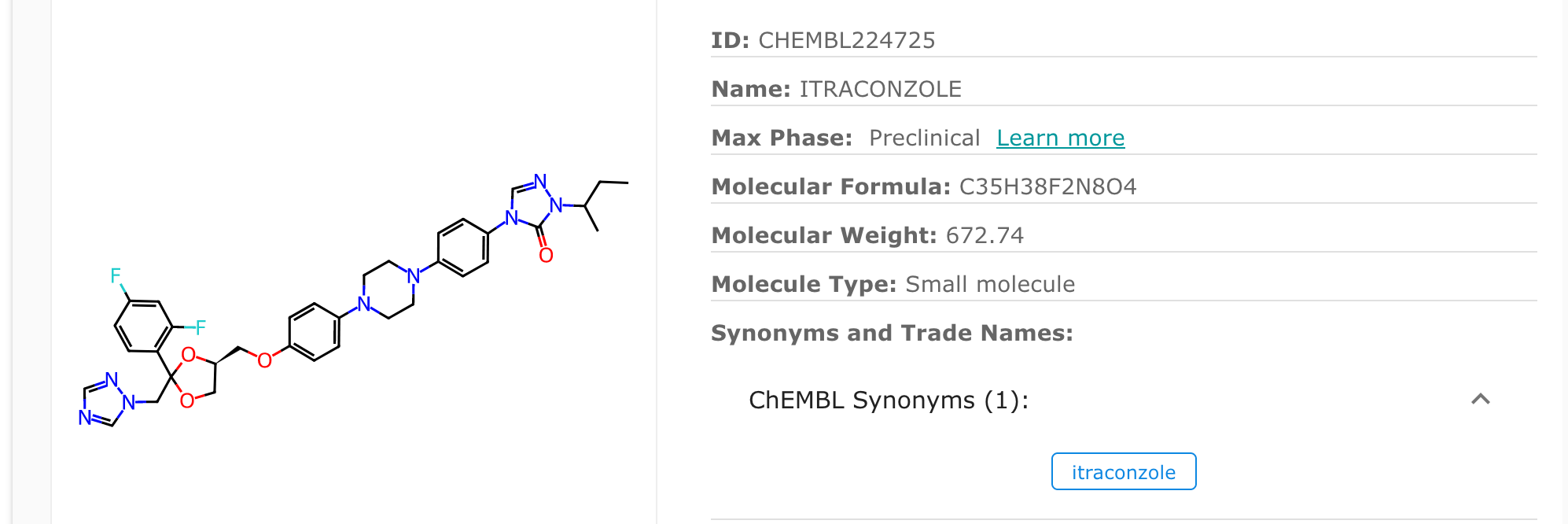
So to add this likely-mis-spelled “itraconzole” into the dataframe, I literally just add it into the SQL query above when obtaining drug information through chembl_downloader.
# SMILES of new addition - "itraconzole"
df_3a4_s_inh_p.loc[6, "canonical_smiles"]'CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6cncn6)(c6ccc(F)cc6F)O5)cc4)CC3)cc2)c1=O'# Labelling stereocentres of new addition - "itraconzole"
itracon_6 = Chem.MolFromSmiles("CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6cncn6)(c6ccc(F)cc6F)O5)cc4)CC3)cc2)c1=O")
rdCIPLabeler.AssignCIPLabels(itracon_6)
itracon_6There is only one stereocentre for “itraconzole”, which will match the CHEMBL224725 entry for “itraconzole” in PubChem. Without looking into other cross-referencing sources, and if only sticking with PubChem for now, I’ve then gone back to check all 3 (stereochemically-)different versions of itraconazole and found that the RDKit stereochemical calculations of these 3 itraconazoles have all basically matched their equivalent PubChem computations for atom sterecentre counts.
CHEMBL22587 - PubChem CID 55283 - 2 defined, 1 undefined atom stereocentre count
itracon_4CHEMBL64391 - PubChem CID 3793 - 0 defined, 3 undefined atom stereocentre count
itracon_5CHEMBL224725 - PubChem CID 44428219 - 1 defined, 2 undefined atom stereocentre count
itracon_6Dataframe df_3a4_s_inh_p (the preprocessed strong CYP3A4 inhibitors) containing 3 different itraconazoles is then updated below to remove two of the triplicated entries.
# Preprocessed df of strong cyp3a4 inhibitors
df_3a4_s_inh_p.head(10)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL2403108 | CERITINIB | 4.0 | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | <rdkit.Chem.rdchem.Mol object at 0x135da19a0> | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | [C][C][=C][C][Branch2][Ring2][=Branch1][N][C][... | InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-... | VERWOWGGCGHDQE-UHFFFAOYSA-N |
| 1 | CHEMBL1741 | CLARITHROMYCIN | 4.0 | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | <rdkit.Chem.rdchem.Mol object at 0x135da1b60> | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | [C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran... | InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21... | AGOYDEPGAOXOCK-KCBOHYOISA-N |
| 2 | CHEMBL2216870 | IDELALISIB | 4.0 | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | <rdkit.Chem.rdchem.Mol object at 0x135da1a80> | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | [C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][... | InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-... | IFSDAJWBUCMOAH-HNNXBMFYSA-N |
| 3 | CHEMBL115 | INDINAVIR | 4.0 | CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](... | <rdkit.Chem.rdchem.Mol object at 0x135da1ee0> | CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](... | [C][C][Branch1][C][C][Branch1][C][C][N][C][=Br... | InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4... | CBVCZFGXHXORBI-PXQQMZJSSA-N |
| 4 | CHEMBL22587 | ITRACONAZOLE | NaN | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](... | <rdkit.Chem.rdchem.Mol object at 0x135da1cb0> | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](... | [C][C][C][Branch1][C][C][N][N][=C][N][Branch2]... | InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2... | VHVPQPYKVGDNFY-ZPGVKDDISA-N |
| 5 | CHEMBL64391 | ITRACONAZOLE | 4.0 | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn... | <rdkit.Chem.rdchem.Mol object at 0x135da1d90> | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn... | [C][C][C][Branch1][C][C][N][N][=C][N][Branch2]... | InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2... | VHVPQPYKVGDNFY-UHFFFAOYSA-N |
| 6 | CHEMBL224725 | ITRACONZOLE | NaN | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6... | <rdkit.Chem.rdchem.Mol object at 0x135da1f50> | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6... | [C][C][C][Branch1][C][C][N][N][=C][N][Branch2]... | InChI=1S/C35H38F2N8O4/c1-3-25(2)45-34(46)44(24... | HUADITLKOCMHSB-RPOYNCMSSA-N |
| 7 | CHEMBL157101 | KETOCONAZOLE | 4.0 | CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c... | <rdkit.Chem.rdchem.Mol object at 0x135da2030> | CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c... | [C][C][=Branch1][C][=O][N][C][C][N][Branch2][R... | InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13... | XMAYWYJOQHXEEK-UHFFFAOYSA-N |
| 8 | CHEMBL45816 | MIBEFRADIL | 4.0 | COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c... | <rdkit.Chem.rdchem.Mol object at 0x135da1e70> | COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c... | [C][O][C][C][=Branch1][C][=O][O][C@][Branch2][... | InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30... | HBNPJJILLOYFJU-VMPREFPWSA-N |
| 9 | CHEMBL623 | NEFAZODONE | 4.0 | CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c... | <rdkit.Chem.rdchem.Mol object at 0x135da20a0> | CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c... | [C][C][C][=N][N][Branch2][Ring1][=Branch2][C][... | InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2... | VRBKIVRKKCLPHA-UHFFFAOYSA-N |
I’m keeping the one with max phase marked as 4.0 (due to the other two having “NaN” with no relevant medical or therapeutic indications data documented in PubChem).
# Note old index unchanged (re-index later if needed)
# Dropping itraconazole at index rows 4 & 6
df_3a4_s_inh_p = df_3a4_s_inh_p.drop(labels = [4, 6])
df_3a4_s_inh_p.head(10)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL2403108 | CERITINIB | 4.0 | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | <rdkit.Chem.rdchem.Mol object at 0x135da19a0> | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | [C][C][=C][C][Branch2][Ring2][=Branch1][N][C][... | InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-... | VERWOWGGCGHDQE-UHFFFAOYSA-N |
| 1 | CHEMBL1741 | CLARITHROMYCIN | 4.0 | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | <rdkit.Chem.rdchem.Mol object at 0x135da1b60> | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | [C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran... | InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21... | AGOYDEPGAOXOCK-KCBOHYOISA-N |
| 2 | CHEMBL2216870 | IDELALISIB | 4.0 | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | <rdkit.Chem.rdchem.Mol object at 0x135da1a80> | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | [C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][... | InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-... | IFSDAJWBUCMOAH-HNNXBMFYSA-N |
| 3 | CHEMBL115 | INDINAVIR | 4.0 | CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](... | <rdkit.Chem.rdchem.Mol object at 0x135da1ee0> | CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](... | [C][C][Branch1][C][C][Branch1][C][C][N][C][=Br... | InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4... | CBVCZFGXHXORBI-PXQQMZJSSA-N |
| 5 | CHEMBL64391 | ITRACONAZOLE | 4.0 | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn... | <rdkit.Chem.rdchem.Mol object at 0x135da1d90> | CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn... | [C][C][C][Branch1][C][C][N][N][=C][N][Branch2]... | InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2... | VHVPQPYKVGDNFY-UHFFFAOYSA-N |
| 7 | CHEMBL157101 | KETOCONAZOLE | 4.0 | CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c... | <rdkit.Chem.rdchem.Mol object at 0x135da2030> | CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c... | [C][C][=Branch1][C][=O][N][C][C][N][Branch2][R... | InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13... | XMAYWYJOQHXEEK-UHFFFAOYSA-N |
| 8 | CHEMBL45816 | MIBEFRADIL | 4.0 | COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c... | <rdkit.Chem.rdchem.Mol object at 0x135da1e70> | COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c... | [C][O][C][C][=Branch1][C][=O][O][C@][Branch2][... | InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30... | HBNPJJILLOYFJU-VMPREFPWSA-N |
| 9 | CHEMBL623 | NEFAZODONE | 4.0 | CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c... | <rdkit.Chem.rdchem.Mol object at 0x135da20a0> | CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c... | [C][C][C][=N][N][Branch2][Ring1][=Branch2][C][... | InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2... | VRBKIVRKKCLPHA-UHFFFAOYSA-N |
| 10 | CHEMBL584 | NELFINAVIR | 4.0 | Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN... | <rdkit.Chem.rdchem.Mol object at 0x135da2180> | Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN... | [C][C][=C][Branch1][C][O][C][=C][C][=C][Ring1]... | InChI=1S/C32H45N3O4S/c1-21-25(15-10-16-28(21)3... | QAGYKUNXZHXKMR-HKWSIXNMSA-N |
| 11 | CHEMBL3545110 | RIBOCICLIB | 4.0 | CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1... | <rdkit.Chem.rdchem.Mol object at 0x135da22d0> | CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1... | [C][N][Branch1][C][C][C][=Branch1][C][=O][C][=... | InChI=1S/C23H30N8O/c1-29(2)22(32)19-13-16-14-2... | RHXHGRAEPCAFML-UHFFFAOYSA-N |
After cleaning up the duplicated structures, below are the full sets of strong and moderate CYP3A4 inhibitors for structural checking.
# strong cyp3a4 inhibitors
Draw.MolsToGridImage(
df_3a4_s_inh_p["rdkit_mol"],
molsPerRow=3,
subImgSize=(400, 300),
legends=list(df_3a4_s_inh_p["pref_name"]))
# moderate cyp3a4 inhibitors
Draw.MolsToGridImage(
df_3a4_m_inh_p["rdkit_mol"],
molsPerRow=3,
subImgSize=(400, 300),
legends=list(df_3a4_m_inh_p["pref_name"])
)
Maximum common substructures
MCS is something I’m interested in trying out so below are some examples of finding MCS in these CYP inhibitors. Please note that MCS may not be the most suitable strategy to look at these CYP inhibitors, I’m only using it to become a bit more familiar with it so that I can better understand MCS.
Some information regarding MCS in RDKit:
FindMCS is for 2 or more molecules and returns single-fragment MCS - based on FMCS algorithm (Dalke and Hastings 2013)
RascalMCES (maximum common edge substructures) is for 2 molecules only and returns multi-fragment MCES. A RDKit blog post by Dave Cosgrove talks about this in more details
Some code examples to refer to:
A RDKit code example may be a good starting point to learn about MCS
TeachOpenCADD’s MCS talktorial - a nice tutorial-like piece that introduces MCS and explains about the FMCS algorithm
“Customising MCS mapping in rdkit” - a more detailed post providing some code examples for atom matching
Strong CYP3A4 inhibitors
I’m starting with the strong CYP3A4 inhibitors first.
# Get list of RDKit mols
mols_s3a4 = list(df_3a4_s_inh_p["rdkit_mol"])
# Find MCS in mols
s3a4_mcs = rdFMCS.FindMCS(mols_s3a4)
# Get images of highlighted MCS for strong CYP3A4 inhibitors
Draw.MolsToGridImage(
mols_s3a4,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_3a4_s_inh_p["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs.queryMol) for m in mols_s3a4]
)
You can get the number of atoms and bonds and also SMARTS string for the MCS like this below.
s3a4_mcs.numAtoms, s3a4_mcs.numBonds(7, 6)s3a4_mcs.smartsString'[#6](-,:[#6](:,-[#7]:,-[#6]):,-[#6])-,:[#6]-,:[#6]'One way to customise MCS is via reducing molecule threshold to relax the MCS rule as suggested by the TeachOpenCADD reference above.
s3a4_mcs_80 = rdFMCS.FindMCS(mols_s3a4, threshold=0.8)
Draw.MolsToGridImage(
mols_s3a4,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_3a4_s_inh_p["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs_80.queryMol) for m in mols_s3a4]
)
# Without changing threshold
s3a4_mcs1 = Chem.MolFromSmarts(s3a4_mcs.smartsString)
# Lowered MCS threshold to 80% of mols
s3a4_mcs2 = Chem.MolFromSmarts(s3a4_mcs_80.smartsString)
Draw.MolsToGridImage([s3a4_mcs1, s3a4_mcs2], legends=["MCS1", "MCS2 with threshold = 0.8"])
Here the MCS differs between different MCS thresholds - when the threshold is not changed, it shows a partial contour of a ring structure, whereas the lowered threshold shows more of an alkyl chain structure.
Moderate CYP3A4 inhibitors
This is then followed by the moderate inhibitors for CYP3A4.
# Get list of RDKit mols
mols_m3a4 = list(df_3a4_m_inh_p["rdkit_mol"])
# Find MCS in mols
m3a4_mcs = rdFMCS.FindMCS(mols_m3a4)
# Get images of highlighted MCS for moderate CYP3A4 inhibitors
Draw.MolsToGridImage(
mols_m3a4,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_3a4_s_inh_p["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(m3a4_mcs.queryMol) for m in mols_m3a4]
)
Aromatic (and macrolide) rings are highlighted in the MCS for this group.
All CYP3A4 inhibitors
The moderate CYP3A4 inhibitors are then added next in order to see if MCS changes when looking at all of the CYP3A4 inhibitors.
# Concatenate dfs for moderate & strong CYP3A4 inhibitors
df_3a4_all = pd.concat([df_3a4_s_inh_p, df_3a4_m_inh_p])
# index un-changed for now
print(df_3a4_all.shape)
df_3a4_all.head(3)(27, 9)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL2403108 | CERITINIB | 4.0 | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | <rdkit.Chem.rdchem.Mol object at 0x135da19a0> | Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c... | [C][C][=C][C][Branch2][Ring2][=Branch1][N][C][... | InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-... | VERWOWGGCGHDQE-UHFFFAOYSA-N |
| 1 | CHEMBL1741 | CLARITHROMYCIN | 4.0 | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | <rdkit.Chem.rdchem.Mol object at 0x135da1b60> | CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(... | [C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran... | InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21... | AGOYDEPGAOXOCK-KCBOHYOISA-N |
| 2 | CHEMBL2216870 | IDELALISIB | 4.0 | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | <rdkit.Chem.rdchem.Mol object at 0x135da1a80> | CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n... | [C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][... | InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-... | IFSDAJWBUCMOAH-HNNXBMFYSA-N |
mols_3a4_all = list(df_3a4_all["rdkit_mol"])
# Find MCS for all CYP3A4 inhibitors
all_3a4_mcs = rdFMCS.FindMCS(mols_3a4_all)
# All CYP3A4 inhibitors
Draw.MolsToGridImage(
mols_3a4_all,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_3a4_all["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs.queryMol) for m in mols_3a4_all]
)
It appears the MCS for most of them involves (partial) rings (e.g. cycloalkane, aromatic and macrolide ones). This result is basically not very different from when we’ve looked at the the CYP3A4 inhibitors separately in their moderate and strong potencies. The next thing I want to try is to add in the ring bonds matching.
# matching ring bonds
all_3a4_mcs_ring = rdFMCS.FindMCS(mols_3a4_all, ringMatchesRingOnly=True)
Draw.MolsToGridImage(
mols_3a4_all,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_3a4_all["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs_ring.queryMol) for m in mols_3a4_all]
)
The ring bond matching also shows a very similar result here as it only restricts the MCS matching to ring bonds only.
Some other interesting code re. MCS
One of the code examples I’d like to try next, in order to see if it makes any differences from above, is the code snippets shared by a RDKit post from Paolo Tosco - “New MCS features in 2023.09.1” - the “Custom Python AtomCompare and BondCompare classes” section.
Some notes from me:
AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in ring systems or
custom subclasses in AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in non-ring systems
i.e. customise parameters using rdFMCS.MCSParameters()
I’ll also attempt to add some code comments below to explain how the code works (anyone’s welcomed to report any issues or changes for this part).
Ring systems
Here, I’m trying the AtomCompare & BondCompare along with RingMatchesRingOnly and CompleteRingsOnly first.
Code
## Customise MCS parameters
# Initiate a MCSParameter object
params = rdFMCS.MCSParameters()
# Define atom typer to be used to compare elements within rdFMCS.AtomCompare class
params.AtomTyper = rdFMCS.AtomCompare.CompareElements
# Define bond typer to be used to compare bond orders within rdFMCS.BondCompare class
params.BondTyper = rdFMCS.BondCompare.CompareOrder
# RingMatchesRingOnly - ring atoms to match other ring atoms only
# CompleteRingsOnly - match full rings only
params.BondCompareParameters.RingMatchesRingOnly = True
params.BondCompareParameters.CompleteRingsOnly = False
all_3a4_ringMCS = rdFMCS.FindMCS(mols_3a4_all, params)
Draw.MolsToGridImage(
mols_3a4_all,
subImgSize=(400, 300),
molsPerRow=3,
legends = list(df_3a4_all["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(all_3a4_ringMCS.queryMol) for m in mols_3a4_all]
)
Some MCS characteristics noted after trying this:
When both BondCompareParameters are set to true, no rings are highlighted (apart from ethyl chains being highlighted in every molecule)
When turning off CompleteRingsOnly, this allows partial rings to be shown in highlighted MCS
A similar output is generated when using ringMatchesRingOnly = True in FindMCS() earlier for all CYP3A4 inhibitors
Non-ring systems
The class code below is also borrowed from the RDKit blog post, which explains why it’s done as a custom “class” code and not a “function” one.
Code
# I've had to think harder about the what the class code are doing below,
# especially the differences between comparing bond orders and ring atoms...
# I can only describe it as both a restrictive (matching non-ring bonds only)
# and lenient (but still comparing ring atoms) process in order to cover the non-ring parts (?)
# Compare bond orders outside ring systems using rdFMCS.MCSBondCompare
# Using class code that will call a function object
class CompareOrderOutsideRings(rdFMCS.MCSBondCompare):
def __call__(self, p, mol1, bond1, mol2, bond2):
# Get bonds 1 and 2 based on bond indices for mols 1 and 2
b1 = mol1.GetBondWithIdx(bond1)
b2 = mol2.GetBondWithIdx(bond2)
# If bonds 1 and 2 are both in ring or if their bond types are the same
if (b1.IsInRing() and b2.IsInRing()) or (b1.GetBondType() == b2.GetBondType()):
# If using stereo matching parameter and not checking bond stereo descriptors,
# return bonds that are not using stereo matching parameter
if (p.MatchStereo and not self.CheckBondStereo(p, mol1, bond1, mol2, bond2)):
return False
# If using ring bonds-matching-ring bonds parameter
# return mols/bonds that are part of a ring only
if p.RingMatchesRingOnly:
return self.CheckBondRingMatch(p, mol1, bond1, mol2, bond2)
return True
# Only match bonds that are not part of ring systems
return False
# Compare atom elements outside ring systems using rdFMCS.MCSAtomCompare
class CompareElementsOutsideRings(rdFMCS.MCSAtomCompare):
def __call__(self, p, mol1, atom1, mol2, atom2):
# Get atoms 1 and 2 based on atom indices for mols 1 and 2
a1 = mol1.GetAtomWithIdx(atom1)
a2 = mol2.GetAtomWithIdx(atom2)
# If atomic numbers for both atoms are different and the atoms are not in ring systems,
# return atoms that instead have the same atomic numbers and in rings systems
if (a1.GetAtomicNum() != a2.GetAtomicNum()) and not (a1.IsInRing() and a2.IsInRing()):
return False
# If using matching atom chirality parameter and not checking both atoms have a chiral tag,
# return atoms that are not using the matching atom chirality parameter
if (p.MatchChiralTag and not self.CheckAtomChirality(p, mol1, atom1, mol2, atom2)):
return False
# If using ring bonds-matching-ring bonds parameter,
# return mols/atoms that are part of a ring only
if p.RingMatchesRingOnly:
return self.CheckAtomRingMatch(p, mol1, atom1, mol2, atom2)
# Only match ring atoms against ring atoms
return True
params_or = rdFMCS.MCSParameters()
params_or.AtomTyper = CompareElementsOutsideRings()
params_or.BondTyper = CompareOrderOutsideRings()
params_or.BondCompareParameters.RingMatchesRingOnly = True
params_or.BondCompareParameters.CompleteRingsOnly = True
all_3a4_orMCS = rdFMCS.FindMCS(mols_3a4_all, params_or)
Draw.MolsToGridImage(
mols_3a4_all,
subImgSize=(500, 400),
molsPerRow=3,
legends = list(df_3a4_all["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(all_3a4_orMCS.queryMol) for m in mols_3a4_all]
)
By using the suggested class code above, the MCS has indeed become larger, where a full ring is highlighted as the MCS.
A second code example from iwatobipen is to highlight molecular differences, which is different from highlighting only the MCSes. Alternative URL link and more examples can be accessed via RDKit Cookbook. Possible use cases of this code may be when dealing with a large set of analogues with changes in R-groups or during large compound screening and search (just as a few examples only). The main thing I can see from the code is that it begins with finding MCS for the input molecules, then uses SMARTS strings of the MCS in order to find atoms not within the MCS (using GetSubstructMatch() part) then highlights that part of the molecules.
All CYP2D6 inhibitors
Because of how the MCSes have turned out for CYP3A4 inhibitors above, I think I should just look at CYP2D6 inhibitors as a whole here. First thing is to combine the dataframes between the moderate and strong inhibitor groups.
# Concatenate dfs
df_2d6_all = pd.concat([df_2d6_s_inh_p, df_2d6_m_inh_p])
# index un-changed for now
print(df_2d6_all.shape)
df_2d6_all.head()(14, 9)| chembl_id | pref_name | max_phase | canonical_smiles | rdkit_mol | standard_smiles | selfies | inchi | inchikey | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | CHEMBL894 | BUPROPION | 4.0 | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | <rdkit.Chem.rdchem.Mol object at 0x135da3530> | CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1 | [C][C][Branch1][#Branch2][N][C][Branch1][C][C]... | InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-... | SNPPWIUOZRMYNY-UHFFFAOYSA-N |
| 1 | CHEMBL41 | FLUOXETINE | 4.0 | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3290> | CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1 | [C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=... | InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-... | RTHCYVBBDHJXIQ-UHFFFAOYSA-N |
| 2 | CHEMBL490 | PAROXETINE | 4.0 | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | <rdkit.Chem.rdchem.Mol object at 0x135da3bc0> | Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1 | [F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@... | InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-... | AHOUBRCZNHFOSL-YOEHRIQHSA-N |
| 4 | CHEMBL1294 | QUINIDINE | 4.0 | C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc... | <rdkit.Chem.rdchem.Mol object at 0x135da2d50> | C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc... | [C][=C][C@H1][C][N][C][C][C@H1][Ring1][=Branch... | InChI=1S/C20H24N2O2/c1-3-13-12-22-9-7-14(13)10... | LOUPRKONTZGTKE-LHHVKLHASA-N |
| 0 | CHEMBL254328 | ABIRATERONE | 4.0 | C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C... | <rdkit.Chem.rdchem.Mol object at 0x135dd0270> | C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C... | [C][C@][C][C][C@H1][Branch1][C][O][C][C][Ring1... | InChI=1S/C24H31NO/c1-23-11-9-18(26)14-17(23)5-... | GZOSMCIZMLWJML-VJLLXTKPSA-N |
mols_all_2d6 = list(df_2d6_all["rdkit_mol"])
# Find MCS for all CYP2D6 inhibitors
all_2d6_mcs = rdFMCS.FindMCS(mols_all_2d6)
# Get images of highlighted MCS for all CYP2D6 inhibitors
Draw.MolsToGridImage(
mols_all_2d6,
subImgSize=(400, 300),
molsPerRow=2,
legends = list(df_2d6_all["pref_name"]),
highlightAtomLists=[m.GetSubstructMatch(all_2d6_mcs.queryMol) for m in mols_all_2d6]
)
Again, only a phenyl ring is highlighted as the MCS so this is not informative at all.
Some findings and future work
Below are some of my small findings and thoughts.
As mentioned at the very top in the outline section, rings (heterocycles, aromatic or fused ones) are everywhere in the MCSes for CYP3A4 and CYP2D6 inhibitors, and they are very common in known approved drugs
Looking at CYP structures in relation to these inhibitors may be more meaningful and also may reveal more insights about possible binding sites or mechanisms of actions of CYP inhibitions for these compounds. This may also be further explored in parallel to the actual drug targets of these CYP inhibitors e.g. binding site on CYP isoenzyme versus binding site on drug target protein as there are different classes of drugs within these CYP inhibitors. For example, some of CYP2D6 inhibitors are drugs acting on central nervous system (about 9 out of 14 drugs) - e.g. bupropion, fluoxetine, paroxetine, clobazam, doxepin, duloxetine, locaserin, moclobemide and rolapitant. For the CYP3A4 inhibitors, they are in several different therapeutic classes e.g. antivirals, antifungals, antibacterials, kinase inhibitors etc.
It may be a bit more interesting to compare the MCSes between CYP3A4 and CYP2D6 substrates (adding in other substrates for other CYPs)
Future posts may involve looking at CYP substrates using different cheminformatics strategies or doing molecular docking within a notebook setting (has this been done before?)
CYP inducers are a different story as they tend to increase drug metabolisms via CYP inductions, which are more likely to do with losing therapeutic effects than gaining adverse effects, so they may be looked at further down the line
MCS may not be absolutely useful in every scenario as I think it aims to look for the most maximum common substructures within a set of molecules, so not every molecule will be able to have a MCS shown (e.g. in a very diverse chemical set), other similarity searching techniques should probably be used instead if needed
Acknowledgements
Thanks to every contributor, developer or authors of every software package used in this post, and also the online communities behind them. Before I forget, the thanks would also be extended to the authors of the journal papers cited in this post. Lastly, special thanks to Noel O’Boyle for being very patient with reading my awfully-long old draft earlier and pointed out some useful things to note and change (I kind of got lost when writing the draft… due to it being a “free-style post”, I should avoid doing this again).
References
Curran, Mark E, Igor Splawski, Katherine W Timothy, G.Michael Vincen, Eric D Green, and Mark T Keating. 1995. “A Molecular Basis for Cardiac Arrhythmia: HERG Mutations Cause Long QT Syndrome.” Cell 80 (5): 795–803. https://doi.org/10.1016/0092-8674(95)90358-5.
Dalke, Andrew, and Janna Hastings. 2013. “FMCS: A Novel Algorithm for the Multiple MCS Problem.” Journal of Cheminformatics 5 (S1). https://doi.org/10.1186/1758-2946-5-s1-o6.
Doveston, Richard G., Paolo Tosatti, Mark Dow, Daniel J. Foley, Ho Yin Li, Amanda J. Campbell, David House, Ian Churcher, Stephen P. Marsden, and Adam Nelson. 2015. “A Unified Lead-Oriented Synthesis of over Fifty Molecular Scaffolds.” Organic & Biomolecular Chemistry 13 (3): 859–65. https://doi.org/10.1039/c4ob02287d.
Flockhart, DA., D. Thacker, C. McDonald, and Z. Desta. 2021. “The Flockhart Cytochrome P450 Drug-Drug Interaction Table.” https://drug-interactions.medicine.iu.edu/.
Fraser, James S., and Mark A. Murcko. 2024. “Structure Is Beauty, but Not Always Truth.” Cell 187 (3): 517–20. https://doi.org/10.1016/j.cell.2024.01.003.
Gaulton, Anna, Anne Hersey, Michał Nowotka, A. Patrícia Bento, Jon Chambers, David Mendez, Prudence Mutowo, et al. 2016. “The ChEMBL database in 2017.” Nucleic Acids Research 45 (D1): D945–54. https://doi.org/10.1093/nar/gkw1074.
Guengerich, F. Peter. 2020. “A History of the Roles of Cytochrome P450 Enzymes in the Toxicity of Drugs.” Toxicological Research 37 (1): 1–23. https://doi.org/10.1007/s43188-020-00056-z.
Jadhav, Ajit, Rafaela S. Ferreira, Carleen Klumpp, Bryan T. Mott, Christopher P. Austin, James Inglese, Craig J. Thomas, David J. Maloney, Brian K. Shoichet, and Anton Simeonov. 2010. “Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease.” Journal of Medicinal Chemistry 53 (1): 37–51. https://doi.org/10.1021/jm901070c.
Taylor, Richard D., Malcolm MacCoss, and Alastair D. G. Lawson. 2014. “Rings in Drugs.” Journal of Medicinal Chemistry 57 (14): 5845–59. https://doi.org/10.1021/jm4017625.